Indication Expansion: What a Scientifically Rigorous Analysis Requires, and What It Can Leave Out
A scientifically rigorous indication expansion workflow should therefore lead with the mechanistic case.

The central question in indication expansion is whether the asset has a credible mechanistic, genetic, and translational basis for changing disease biology in the proposed patient population. Commercial attractiveness in a new disease area says nothing about that basis on its own.
That distinction sounds obvious, but it is where many indication expansion analyses begin to lose scientific discipline.
An approved or clinical-stage asset brings real strategic advantages including known pharmacology, existing exposure data, a development history, and often a plausible reason to look beyond the initial indication. Those advantages can make adjacent disease opportunities appear compelling. But the existence of an opportunity is not the same as the existence of a biological argument. What transfers to the new indication is the asset’s pharmacology, exposure, and safety history. The causal disease hypothesis does not transfer with it and must be re-established for each target-indication pair rather than inherited from the original program.
For R&D leaders, the first-order question should be whether perturbing the asset's target, pathway, cell type, or biological process is plausibly causal in the candidate indication. Human genetic evidence is particularly valuable because naturally occurring variation can function as a human perturbation experiment, helping distinguish causal biology from reactive association. The analogy has limits, though. Germ line variants model chronic, low-magnitude perturbation rather than acute pharmacological intervention in established disease, and development compensation can widen that gap further.
Separately, because association data may typically skew heavily toward European-ancestry cohorts, genetic support for a target may not generalize to the proposed patient population. Recent work estimated that drug mechanisms with genetic support have a 2.6-fold higher probability of clinical success than those without, although the strength of that relationship depends on confidence in the causal gene and the quality of the underlying evidence.
A scientifically rigorous indication expansion workflow should therefore lead with the mechanistic case. It should ask whether the disease biology overlaps with the asset biology, whether the relevant patient population expresses or depends on that biology, and whether the clinical evidence supports the proposed causal path.
Indication expansion efforts typically fail from unstructured evidence, information that arrives fragmented, inconsistently weighted, and disconnected from a decision, even when access to that information was never the constraint
The typical pattern is familiar. One team gathers literature, while another reviews genetics or may look at clinical precedent. Someone else may bring in epidemiology, safety context, or competitive activity. Each domain may be handled competently, but the synthesis depends heavily on individual expertise, local habits, and the person who happens to be assembling the final narrative.
The result is a manual stitching exercise, where evidence is retrieved without being consistently reconciled, and contradictions are noted but not always interpreted. Outputs may be comprehensive, yet difficult to compare across hypotheses because they vary in structure, depth, and evidentiary standards. Ranking then begins to feel subjective, not necessarily because the scientists are subjective, but because the workflow has not made the underlying criteria explicit.
Indication expansion is fundamentally a synthesis, comparability, and governance problem, and that is the organizational pain point that deserves more attention.
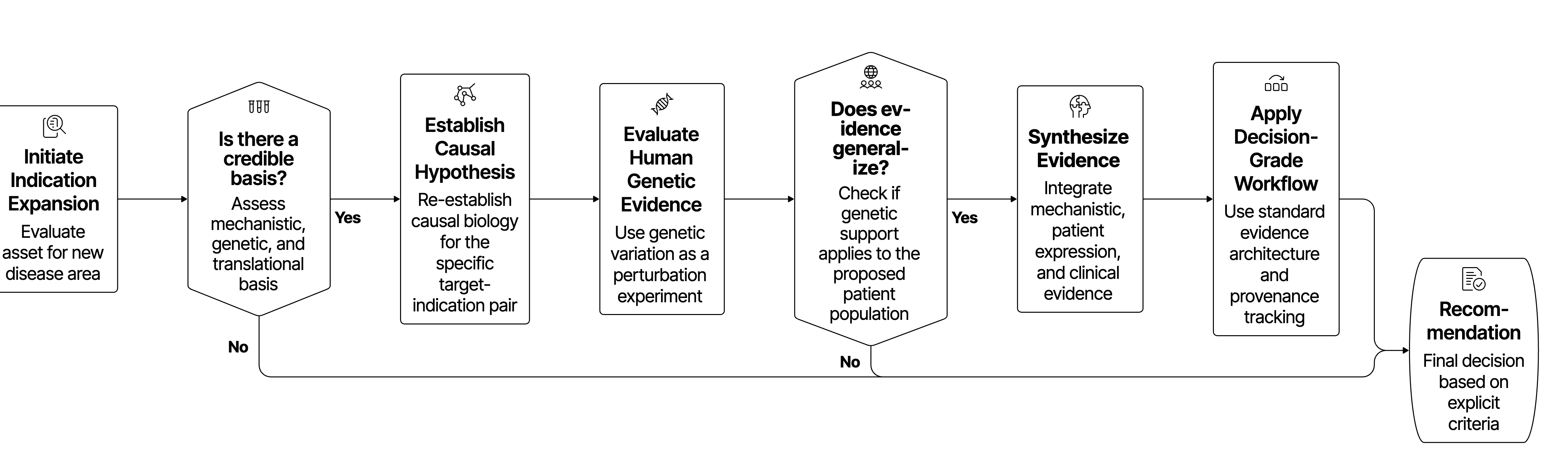
A decision-grade workflow must reduce that variability. It needs a standard evidence architecture, a visible hierarchy of evidence, a definition of what "complete enough" means at each stage, and a way to preserve provenance from source evidence to recommendation. Without those elements, the same evidence landscape can produce different conclusions depending on who searched, which sources were emphasized, and how the story was assembled.
A rigorous workflow should move through three gates
A useful way to structure indication expansion is as a sequence of decision gates, each with a different scientific burden.
Biology is iterative, and new evidence can and should send a team back to earlier assumptions, so the gate structure does not impose artificial linearity. It makes the decision logic inspectable instead, separating early plausibility from deeper biological rationale, and both from portfolio commitment.
Gate 1: Mechanistic plausibility
The first gate asks a narrow question: is the mechanism strong enough to justify looking beyond the current or primary indication?
At this stage, the workflow should focus on the asset's mechanism of action, target biology, pathway directionality, tissue and cell context, and early evidence that the target or pathway intersects disease-relevant biology, with human genetics, perturbation data, pathway biology, and disease pathology dominating the analysis.
The key requirement is directionality. The proposed pharmacology must align with the direction of disease biology, beyond a simple association between target and disease. A protective loss-of-function signal may support inhibition, while a disease-causing loss-of-function signal may argue against inhibition and potentially support agonism, replacement, or pathway compensation. A pathway that is elevated in disease may be pathogenic, compensatory, or merely reactive. Preserving those distinctions makes a mechanistic hypothesis testable instead of merely plausible.
This is also where negative evidence should appear early such as: genetic null findings in well-powered datasets, failed perturbation experiments, lack of pathway activation in the relevant tissue, or evidence that the biology is downstream of disease rather than causal. These findings can all materially weaken the expansion hypothesis, though not all carry equal weight. A failed trial of a related mechanism functions as strong disconfirmation, while a genetic null carries weaker, conditional weight. Absence of association can reflect mutational constraint, allele frequencies below detection, or insufficient power instead of absence of causal involvement.
The weight given to negative evidence should track its interpretability, since treating all negative findings as uniformly decisive overstates their certainty. Drug-target Mendelian randomization and related causal-inference approaches add useful evidence here, but they require careful attention to instrument validity, tissue relevance, pleiotropy, and whether the genetic perturbation approximates the pharmacology of the asset.
The output of this gate should not be a mechanistic rationale artifact containing the proposed causal chain, the strongest supporting evidence, the strongest counterevidence, and a clear go, no-go, or hold position for further exploration, in place of an open-ended literature review.
Gate 2: Biological rationale
The second gate asks which diseases are plausible candidates given the mechanism, and which subset merits deeper evaluation.
This is where indication expansion often becomes analytically overloaded. A mechanism may touch many diseases, phenotypes, and pathways. Without structure, the workflow can generate a longlist that looks impressive but is difficult to interpret.
The better approach is to cluster candidate diseases by shared biological profile, including pathway activation, cell type, tissue context, genetic architecture, inflammatory or fibrotic phenotype, biomarker profile, and mechanistic proximity to the asset, in place of grouping by therapeutic area or clinical label alone.
This is because disease labels are often too coarse to support expansion decisions. A broad indication may contain multiple molecularly distinct patient subsets, so the relevant question becomes whether the patients likely to be treated have the biology the asset is designed to modulate, beyond a general association between target and disease.
Regulatory guidance on enrichment strategies reflects the same underlying principle that clinical development can be strengthened by identifying patients more likely to demonstrate the relevant drug effect, whether through prognostic, predictive, or practical enrichment approaches.
At this gate, the workflow should produce a shortlist rather than a ranked list with unexplained scores. The shortlist should show why each candidate disease is included, why others were excluded, what evidence drives confidence, and which assumptions are carrying the most weight.
The workflow should also make novelty defensible at this gate. A non-obvious indication is valuable only if the evidence chain is traceable. Otherwise, novelty can become indistinguishable from noise.
Gate 3: Prioritization and commitment
The third gate asks which of the shortlisted indications should be prioritized for investment, and why.
At this point, biology remains central, but it is no longer sufficient on its own. A decision-grade assessment must integrate clinical feasibility, endpoint precedent, safety compatibility, patient unmet need, epidemiology, and competitive context. Sequencing matters here, since these dimensions should refine or challenge the mechanistic hypothesis, instead of obscuring its weakness.
Clinical feasibility should assess whether the biology can be tested in a plausible development path. Are there clinically meaningful endpoints? Are biomarkers available or developable? Is the patient population identifiable? Is disease progression measurable in a timeframe compatible with development? Is there precedent for regulatory acceptance of endpoints or enrichment strategies?
Safety compatibility should be indication-specific. A safety liability that is unacceptable in a mild chronic disease may be more tolerable in a severe disease with high unmet need. Conversely, a mechanism that looks attractive biologically may become a risk if the relevant patient population is vulnerable to the predicted adverse event profile. A single generic safety bar is rarely adequate. The on-target, mechanism-based safety read should already exist from Gate 1; what is indication-specific here is the level of risk that the population and unmet need will tolerate, distinct from the underlying liability.
Competitive intelligence belongs here when it informs mechanism, precedent, differentiation, or feasibility. It is less useful when it becomes an exhaustive landscape detached from the biological question.
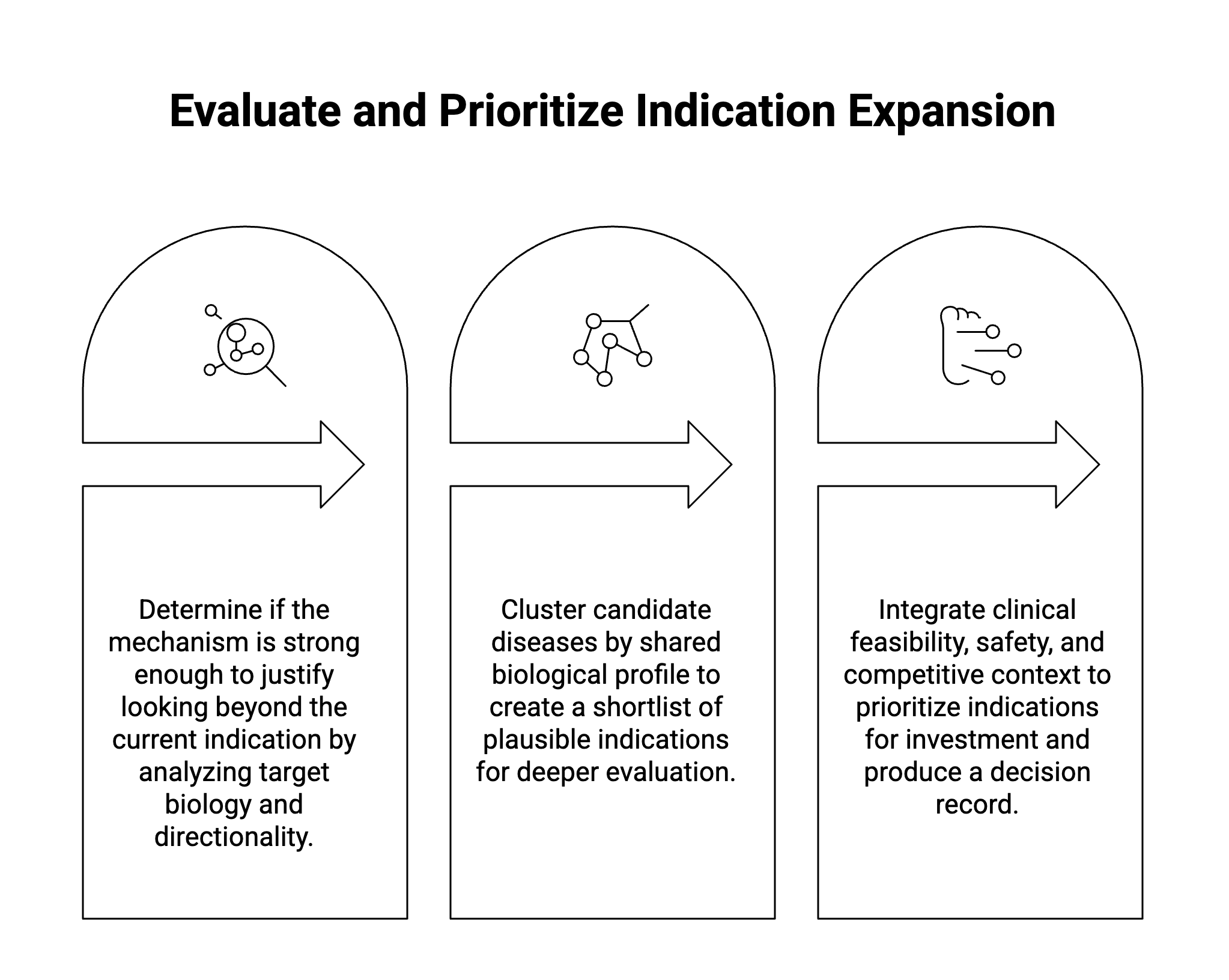
The output should be a decision record for that specific candidate, containing the recommendation, the evidence supporting it, the counterarguments, the key uncertainties, and the specific data that would change the conclusion.
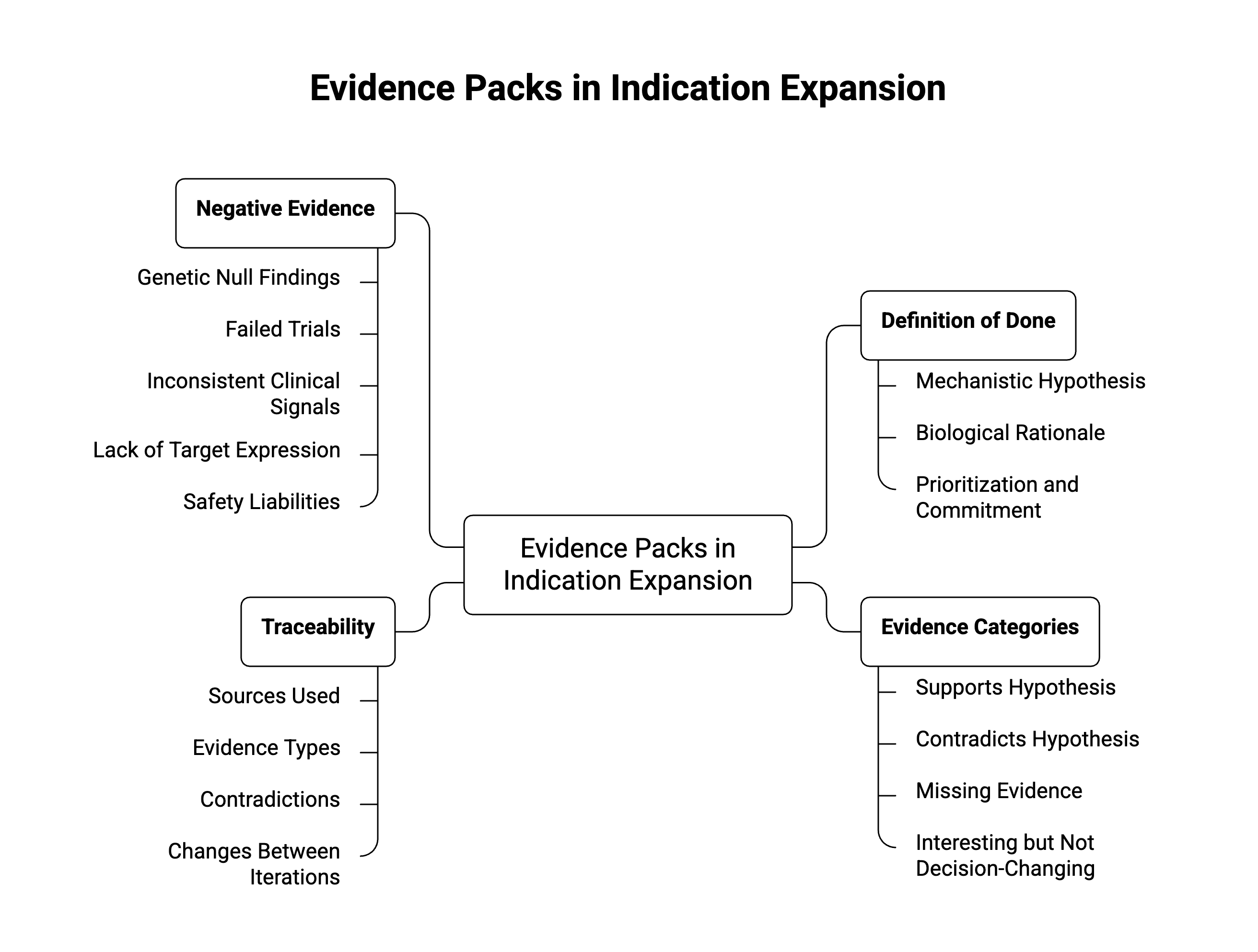
Evidence packs need a definition of done
One of the more persistent pain points in indication expansion is the absence of a shared bar for when an analysis is complete enough to support the next decision.
This does not mean every workflow should demand perfect evidence, since that would be unworkable given that early indication expansion often proceeds under uncertainty. But that uncertainty should be structured instead of left unexamined.A useful evidence pack should distinguish four categories:
- Evidence that supports the mechanistic hypothesis.
- Evidence that contradicts or weakens it.
- Evidence that is missing but material to the decision.
- Evidence that is interesting but not decision-changing.
That fourth category is important. Many indication expansion outputs become bloated because they treat all missing information as equally important. A rigorous workflow should be selective, prioritizing the gaps that would change the recommendation over those that additional time could simply fill. Clinical evidence, in particular, should be graded for strength and proximity to the mechanism and labeled plainly as strong, limited, or absent, so that confidence is not implied where the evidence is thin or indirect.
Traceability matters here as well. Senior reviewers need to know not only what the workflow concluded, but how it got there. Which sources were used? Which evidence types carried the most weight? Were contradictions found? Were they resolved, discounted, or left unresolved? What changed between one iteration and the next?
Without that provenance, teams may trust the author but not the result. That distinction becomes costly when work moves across functions, programs, or governance bodies.
Negative evidence should be designed into the workflow
A credible indication expansion workflow should actively search for evidence against the hypothesis.
That includes genetic null findings, failed trials of related mechanisms, inconsistent clinical signals, pathway biology that suggests compensation instead of causation, lack of target expression in relevant tissue, or safety liabilities that are especially problematic in the proposed population.
The published literature is not an unbiased representation of all scientific results. Publication bias and outcome reporting bias can make positive or statistically significant findings easier to retrieve than negative or non-significant findings, which means a workflow that only gathers supportive evidence may create a falsely coherent picture.
This is especially risky in indication expansion, where teams may already have a strategic reason to want the hypothesis to work. The risk is sharpest in due-diligence and portfolio-review settings, where a one-sided read can drive an investment decision. A workflow that produces only supportive evidence may feel efficient, but it is not necessarily rigorous. The counterargument is part of the scientific product.
The goal is to calibrate confidence by weighing negative evidence on its merits, without defaulting every recommendation toward caution. Some expansion hypotheses will remain compelling after negative evidence is considered. Those are the ones worth pursuing further.
What can be left out
A scientifically focused indication expansion workflow does not need to include everything. The dimensions Gate 3 introduces, such as epidemiology and competitive context, earn their place only when they bear on the mechanistic or feasibility argument; where they do not, they may be the first candidates for exclusion.
Exhaustive epidemiological background can be left out unless it affects patient selection, feasibility, disease segmentation, or unmet need. And the same applies to detailed market sizing, which the scientific assessment unless it is needed to define the population under consideration. Broad competitive intelligence follows the same logic, warranting inclusion only when competitor mechanisms provide mechanistic precedent, clinical validation, safety warnings, endpoint precedent, or differentiation constraints.
Those topics are often essential for portfolio strategy and business development. They simply answer a different question than the scientific one.
A scientific indication expansion workflow asks whether the biological hypothesis is credible and testable. A commercial assessment asks whether the opportunity is attractive and executable. Conflating those two decisions can weaken both. A weak mechanistic case should not be rescued by a large market just as a strong mechanistic case should not be dismissed prematurely because the commercial analysis belongs in a later, separate discussion.
The cleanest workflow keeps the mechanistic argument visible, then layers clinical, safety, epidemiological, and competitive considerations in proportion to their decision relevance.
The workflow output should arrive at a position
An indication expansion workflow should conclude in a scientific argument that takes a position on the evidence, distinct from a dossier that simply compile it.
That argument can be qualified. It might conclude that the mechanistic case is plausible but not yet human-validated, that the genetic evidence is supportive but directionally ambiguous, or that the clinical precedent is weak, but a biomarker strategy could still make the hypothesis testable. It might equally conclude that the opportunity should not progress, because the strongest available evidence argues against causal involvement of the target.
It should not return the decision to the reviewer as a comprehensive but noncommittal evidence summary.A decision-grade output must take a position, surface the contradictions that matter, and name the gaps that would change the conclusion. That is what allows portfolio committees, translational leaders, and governance bodies to evaluate the science rather than reconstruct the analysis themselves.
Scientific credibility comes from the structure of the reasoning
Indication expansion is attractive because it appears to offer leverage, including known pharmacology, existing development history, and the possibility of extending value into new patient populations. But that same attractiveness creates a risk of confirmatory analysis.
The antidote is workflow design.
A rigorous indication expansion workflow is first grounded with biological mechanistic insight, and causal human evidence over associative evidence. It asks whether the intended patients have the relevant biology, grades clinical evidence by strength and proximity to the mechanism. It incorporates safety and feasibility early enough to influence prioritization, preserves provenance, and gives negative evidence enough weight to change the conclusion.
The purpose is to make the few hypotheses that are truly promising easier to distinguish from those that are only superficially attractive.
The gate structure above only holds up if the underlying evidence stays traceable from source to recommendation, which is the harder infrastructure problem that most teams have not solved. Evaluate whether your current indication expansion process can show, for any recommendation, which evidence supported it, which evidence weighed against it, and what would change the conclusion.
See how Causaly's Scientific Workflows preserves that chain across mechanistic, genetic, and clinical evidence so the reasoning stays inspectable at every gate.
Further reading


.png)

.png)
.png)


Get started with Causaly
Ready to transform the way your R&D teams discover and deliver? Take the first step - see Causaly for yourself.
Request a demo